Drug metabolism (biotransformation) is a process by which the body converts drugs and pharmaceuticals into compounds that are more easily water-soluble for elimination by the kidneys and liver.
The body employs three sequential phases to achieve complete drug elimination:
- Phase I: Oxidation, reduction, and hydrolysis—expose or create functional groups
- Phase II: Conjugation reactions—attach polar groups to increase water solubility
- Phase III: Active transport—pumps drugs and metabolites across membranes for elimination
| Feature | Phase I (Functionalization) | Phase II (Conjugation) | Phase III (Elimination) |
| Primary Goal | Expose/create a functional “handle” (e.g., -OH, -NH2). | Covalently attach a polar group to the “handle”. | Active transport of metabolites out of the cell. |
| Major Reactions | Oxidation, Reduction, Hydrolysis. | Glucuronidation, Acetylation, Sulfation, GSH conjugation. | Efflux via ATP-dependent pumps. |
| Key Enzymes | Cytochrome P450 (CYP450). | UGT, NAT, SULT, GST. | P-glycoprotein (MDR1), OATP. |
| Result | Metabolite may be active (prodrug) or toxic. | Almost always results in an inactive, polar metabolite. | Removal from the body via bile or urine. |
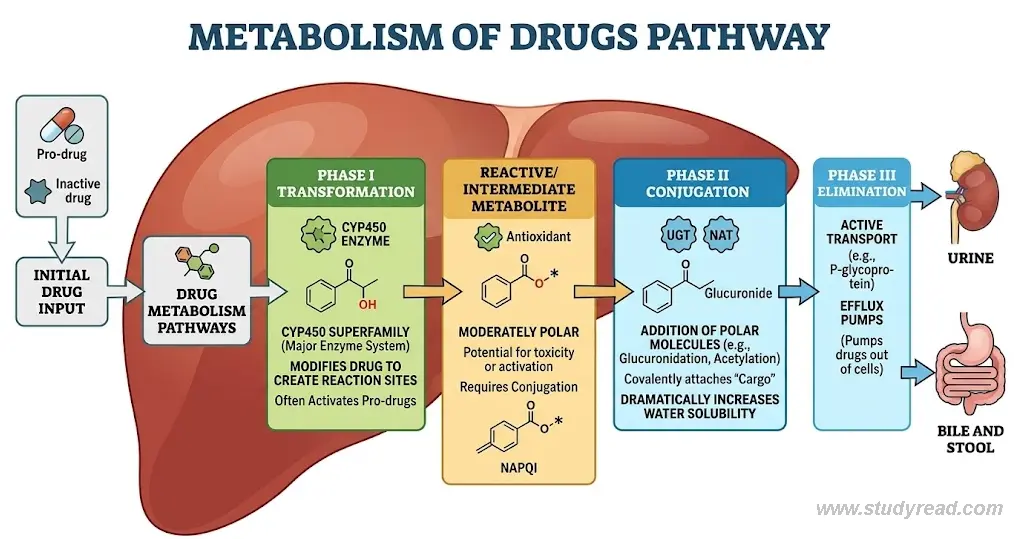
Understanding these pathways helps health care professionals to predict drug interactions, toxicity, and individual variability in drug response and optimize clinical therapy.
Besides, this knowledge is essential for medical board exams.
Phase I Metabolism: Oxidation, Reduction, and Hydrolysis
Phase I reactions are primarily catalyzed by the cytochrome P450 (CYP450) enzyme system in hepatic cells.
These reactions modify the drug’s molecular structure, exposing or creating functional groups. Thus, these newly formed metabolites have greater water solubility than the parent molecule or serve as substrates for Phase II conjugation reactions.
These phase I reactions are of three types, such as
- Oxidation
- Reduction
- Hydrolysis
Oxidation (Primary Phase I Reaction)
This is the most common reaction of the Phase I pathway and involves the addition of oxygen or the removal of hydrogen.
The reaction is catalyzed by CYP450 using NADPH and molecular oxygen:
R-H + O₂ + NADPH → R-OH + H₂O + NADP⁺
Common oxidative pathways include:
- Aromatic hydroxylation: benzene → phenol (creates hydroxyl group on aromatic ring)
- Aliphatic hydroxylation: propranolol → 4-hydroxypropranolol (oxidation of side chain)
- N-dealkylation: codeine → morphine (removes methyl group from nitrogen)
- O-dealkylation: acetaminophen → N-acetyl-p-benzoquinone imine (NAPQI, reactive intermediate)
- Deamination: amphetamine → phenylacetone (removes amino group)
Reduction
Reduction reaction involves the addition of hydrogen or the removal of oxygen from drug molecules.
Mostly carbonyl compounds are involved and are catalyzed by aldehyde dehydrogenase, alcohol dehydrogenase, and ketone reductases.
R-C=O + NADPH + H⁺ → R-CHOH + NADP⁺
- Ketone reduction: corticosteroids → less active alcohols
- Aldehyde reduction: disulfiram metabolites (reduced by aldehyde dehydrogenase)
Hydrolysis
Hydrolysis reaction involves breaking ester and amide bonds by the addition of water, catalyzed by enzymes such as esterases and proteases, producing carboxylic acids and alcohols.
This reaction is especially important for prodrugs:
R-COOR’ + H₂O → R-COOH + R’-OH
- Ester hydrolysis: enalapril → enalaprilat (ACE inhibitor prodrug activation)
- Amide hydrolysis: procainamide → p-aminobenzoic acid (PABA, antigenic metabolite)
- Lactone hydrolysis: irinotecan → SN-38 (active topoisomerase inhibitor)
The Cytochrome P450 System: Key Players
CYP450 isoenzymes comprise ≈57 human enzymes. These three isoenzymes metabolize ~80% of all drugs:
| Enzyme | % of Drugs Metabolized | Key Substrates | High-Yield Clinical Fact |
|---|---|---|---|
| CYP3A4 | >50% | Statins, Ca²⁺ blockers, protease inhibitors, macrolides, cyclosporine | Inhibited by grapefruit juice and induced by rifampin. |
| CYP2D6 | ~20% | Codeine, tramadol, β-blockers, antipsychotics, antiarrhythmics | Highly Genetic polymorphisms → ultra-rapid to poor metabolizers; prodrug activation critical. |
| CYP2C19 | ~10% | Warfarin, NSAIDs, clopidogrel, PPIs, phenytoin | Critical for activating the prodrug clopidogrel; “poor metabolizers” risk stent thrombosis. |
While Oxidation, Reduction, and Hydrolysis are the primary Phase I pathways, other specialized reactions, such as decarboxylation, are also clinically essential for activating specific prodrugs, such as Levodopa.
Phase II Metabolism: Conjugation Reactions
Phase II reactions attach highly polar molecules to drugs or Phase I metabolites, dramatically increasing water solubility and facilitating renal/biliary excretion.
These are typically synthetic reactions requiring ATP and enzyme catalysis.
| Pathway | Enzyme | Example Substrate | Board Review “Pearl” |
|---|---|---|---|
| Glucuronidation | UGT | Acetaminophen, Morphine | The most common Phase II reaction. |
| Acetylation | NAT | Isoniazid, Procainamide | Slow Acetylators (NAT2) face a higher risk of drug-induced lupus. |
| GSH Conjugation | GST | NAPQI (Toxic) | Protects the liver from acetaminophen-induced necrosis. |
| Sulfation | SULT | Minoxidil | Necessary to activate minoxidil for hair regrowth. |
| Methylation | MET | Isoproterenol | Minor pathway |
Glucuronidation (UDP-Glucuronosyltransferase, UGT)
Glucuronidation is the most abundant Phase II pathway and transfers glucuronic acid from UDP-glucuronic acid (UDPGA) to nucleophilic groups on drugs:
Drug-OH + UDPGA → Drug-O-glucuronide + UDP
- O-glucuronidation: morphine → morphine-3-glucuronide (analgesic metabolite), acetaminophen → acetaminophen-glucuronide
- N-glucuronidation: lamotrigine → lamotrigine-glucuronide
- Ether glucuronidation: naproxen → naproxen glucuronide
Acetylation (N-Acetyltransferase, NAT)
Acetylation transfers an acetyl group from acetyl-CoA to amines or amides. NAT1 and NAT2 are the primary human isoforms. The rate of acetylation is genetically determined:
Drug-NH₂ + Acetyl-CoA → Drug-NH-COCH₃ + CoA
Acetylated drugs include isoniazid, procainamide, sulfasalazine, sulfonamides, and hydralazine. The NAT2 gene exists in two functional variants:
- Fast acetylators: rapid drug metabolism, lower drug levels
- Slow acetylators: impaired metabolism, higher drug accumulation, toxicity risk
CLINICAL PEARL: NAT2 Polymorphism & Slow Acetylators
- Prevalence: 50% Caucasians, 10-20% East Asians
- Clinical consequence: Slow acetylators of isoniazid (TB prophylaxis) face increased risk of peripheral neuropathy due to drug accumulation. Similarly, procainamide accumulation causes drug-induced lupus (anti-histone antibodies).
- Board tip: When a patient on isoniazid develops neuropathy, suspect slow acetylator phenotype—check NAT2 genotype and consider dose adjustment.
Sulfation (Sulfotransferase, SULT)
Sulfation transfers a sulfate group (SO₄²⁻) from 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to hydroxyl or amino groups. Sulfation is especially important for phenolic and alcoholic compounds:
Drug-OH + PAPS → Drug-O-SO₃⁻ + PAP
- Phenolic sulfation: acetaminophen → acetaminophen sulfate (alternative to glucuronidation)
- Amine sulfation: minoxidil → minoxidil sulfate (active form for hair regrowth)
Glutathione Conjugation (Glutathione S-Transferase, GST)
Glutathione conjugation involves the tripeptide glutathione (γ-glutamyl-cysteinyl-glycine) attacking electrophilic sites on drugs via glutathione S-transferase. This pathway is critical for detoxifying reactive metabolites:
Drug-electrophile + GSH → Drug-S-glutathione conjugate
- Acetaminophen (NAPQI detoxification): NAPQI + GSH → acetaminophen-glutathione (harmless, excreted); overdose depletes GSH → NAPQI accumulates → hepatotoxicity
- Chemotherapy: Alkylating agents (nitrogen mustards, cisplatin) conjugated with GSH—elevated GST expression in tumors confers drug resistance.
Methylation
- It plays a minor role in drug metabolism but is essential for the biosynthesis of endogenous compounds such as epinephrine.
- In this reaction, a methyl group is added to the metabolite, forming a conjugate.
- Enzyme methyltransferase helps in the conjugation.
Phase I vs. Phase II Summary
| Characteristic | Phase I (CYP450) | Phase II (Conjugation) |
|---|---|---|
| Reaction Type | Oxidation, reduction, hydrolysis—modifies structure | Glucuronidation, acetylation, sulfation, glutathione conjugation—attaches polar groups |
| Primary Enzyme | Cytochrome P450 (CYP3A4, 2D6, 2C9, 2C19) | UGT, NAT, SULT, GST |
| Cofactor/Substrate | NADPH, O₂ | UDPGA, Acetyl-CoA, PAPS, GSH |
| Polarity Increase | Moderate (may require Phase II) | Highly polar (readily excreted) |
| Location | Hepatic smooth ER; GI, kidney, lung | Hepatic cytoplasm/ER; extrahepatic |
Phase III: Drug Transport and Elimination
- Phase III involves active, ATP-dependent transport of drugs and metabolites across membranes via carrier proteins.
- These transporters determine whether drugs remain intracellular or are pumped out for elimination.
Two main transporter types:
- Uptake Transporters
- Efflux Transporters
Uptake Transporters (Organic Anion Transporting Polypeptides, OATP)
- OATP1B1 and OATP1B3 facilitate the active uptake of drugs from portal blood into hepatocytes.
Without this transporter-mediated uptake, drugs cannot undergo Phase I metabolism or be excreted into bile:
- Substrate drugs: Statins, bosentan, rifampicin, glucuronidated metabolites
- Genetic variants: OATP1B1 polymorphisms reduce statin uptake → elevated LDL cholesterol, reduced statin efficacy
- Drug interactions: Ketoconazole inhibits OATP → reduces atorvastatin uptake → elevated statin levels → increased myopathy risk
Efflux Transporters (P-Glycoprotein, MDR1/ABCB1)
- P-glycoprotein is an ATP-dependent pump that expels drugs from the cells, reducing their intracellular concentrations and bioavailability.
- It is located on hepatocytes, intestinal enterocytes, renal tubules, and, importantly, the blood-brain barrier (BBB):
- Mechanism: Binds drug substrates intracellularly and pumps them back out using ATP hydrolysis. Works bidirectionally at intestinal enterocytes (reduces oral bioavailability) and at the BBB (prevents CNS penetration).
- Common substrates: Digoxin, fexofenadine, dabigatran, antiretrovirals, some anticancer drugs
- Inducers/inhibitors: Rifampin induces P-gp (reduces drug levels); verapamil, ketoconazole inhibit P-gp (increases drug levels)
CLINICAL PEARL: Grapefruit & Drug Interactions
- Mechanism: Grapefruit juice contains furanocoumarins that irreversibly inhibit intestinal CYP3A4 AND P-glycoprotein. Single glass of grapefruit juice → 2-3 hour effect on CYP3A4; lasts 24+ hours for P-gp.
- Clinical consequences: Simvastatin oral bioavailability increases ↑16-fold → rhabdomyolysis risk. Tacrolimus levels ↑3-4 fold → nephrotoxicity. Fexofenadine ↓50% → reduced antihistamine effect.
- Board tip: Patient on simvastatin presents with rhabdomyolysis → ask about citrus juice. If taking tacrolimus with elevated trough levels → counsel against grapefruit.
CLINICAL PEARL: CYP2C19 Polymorphism & Clopidogrel Non-Responders
- Prodrug mechanism: Clopidogrel is a PRODRUG that requires CYP2C19-mediated oxidation (Phase I) to generate its active thiol metabolite, which irreversibly inhibits P2Y12 platelet receptors.
- Loss-of-function alleles: CYP2C19*2 and *3 reduce enzyme activity by 50-80%. Loss-of-function allele carriers (14-35% of populations, depending on ancestry) generate insufficient active metabolite.
- Clinical consequences: Poor metabolizers have ↓ platelet inhibition (diminished antiplatelet effect), ↑ stent thrombosis risk post-PCI, and recurrent acute coronary syndromes
- Board tip: Patient presents with stent thrombosis within 1 year of PCI on clopidogrel → suspect CYP2C19 loss-of-function allele. Consider genetic testing and switching to prasugrel or ticagrelor (not affected by CYP2C19). The FDA recommends testing before clopidogrel initiation in high-risk patients.
Enzyme Induction and Inhibition: Drug-Drug Interactions
- Understanding CYP450 induction and inhibition is critical for predicting drug-drug interactions, dosing adjustments, and treatment failures.
CYP3A4 Inhibition
Inhibition reduces drug metabolism → increases substrate drug levels → toxicity risk. Inhibition occurs within 1-2 days and reverses in 2-7 days after discontinuation.
- Strong inhibitors: Ketoconazole, itraconazole, ritonavir, clarithromycin
- Moderate inhibitors: Diltiazem, verapamil, amiodarone, fluconazole
- Clinical example: Ketoconazole + simvastatin → simvastatin levels ↑16-fold → rhabdomyolysis
CYP3A4 Induction
Induction increases enzyme expression → accelerates metabolism → reduces substrate levels → therapeutic failure. Induction requires 5-7 days, and reversal takes 1-2 weeks.
- Strong inducers: Rifampin, phenytoin, carbamazepine
- Mild inducers: St. John’s wort, phenobarbital
- Clinical example: Rifampin + warfarin → warfarin levels ↓50-70% → subtherapeutic INR despite adequate dose
CLINICAL PEARL: Rifampin—The Prototypical CYP450 Inducer
- Mechanism: Rifampin activates pregnane X receptor (PXR), upregulating expression of CYP3A4, CYP2C9, CYP2C19, and UDP-glucuronosyltransferase genes. The effect begins in 2-3 days and peaks at 7-10 days.
- Substrates affected: Warfarin (↓INR), tacrolimus (↓drug levels), oral contraceptives (breakthrough bleeding/unintended pregnancy), verapamil, clarithromycin, statins
- Particularly dangerous combinations: Oral contraceptives + rifampin = pregnancy risk. Warfarin + rifampin = need for 2-3× normal warfarin dose. Avoid in heart transplant patients on calcineurin inhibitors.
- Board tip: Female on oral contraceptives starting TB prophylaxis with rifampin → counsel on backup contraception. Patient on warfarin + rifampin → expect INR to drop within 3-5 days; anticipate need for higher warfarin maintenance dose.
Prodrugs: Activation via Phase I Metabolism
Prodrugs are inactive compounds that require Phase I metabolism to convert into an active metabolite. Genetic or pharmacological impairment of the relevant enzyme abolishes drug efficacy.
- Codeine (CYP2D6 prodrug): O-demethylation → morphine. Ultra-rapid metabolizers generate excessive morphine (toxicity); poor metabolizers get no analgesia.
- Tramadol (CYP2D6 prodrug): N-demethylation → active metabolites. Poor metabolizers have reduced efficacy; CYP2D6 inhibitors (e.g., fluoxetine, paroxetine) reduce tramadol’s effect.
- Enalapril (esterase prodrug): Hepatic esterase-mediated hydrolysis → enalaprilat (active ACE inhibitor). Patients with severe liver disease have impaired activation; use lisinopril instead.
- Clopidogrel (CYP2C19 prodrug): Discussed above—loss-of-function alleles eliminate efficacy.
References
- Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 14th ed. McGraw-Hill; 2022. Chapters 1-3 (ADME).
- FDA Guidance for Industry: Clinical Drug Interaction Studies—Cytochrome P450-Mediated Drug Interactions.
- Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine.
- Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition
